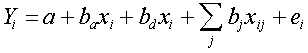|
|
|
|
|
|
|
|
|
|
||
|
RESEARCH PLAN |
|||
|
Principal Investigator/Program Director Williams, Robert W. |
|||
Status of Current ResearchBackgroundQTL mapping includes three levels of increasing complexity. First is single-locus mapping: statistical tests of association between trait values and the genotypes of marker loci throughout the genome. A significant association is interpreted as indicating the presence of a QTL linked to the marker that shows the association. Since these tests consider only one locus at a time, they do not require any information about adjacent loci; that is, they do not require that marker loci be mapped. The second method, simple interval mapping (SIM), requires construction of a marker genetic map. This method evaluates the association between the trait values and the expected genotype of a hypothetical QTL (the target QTL) at multiple analysis points between each pair of adjacent marker loci (the target interval). The expected QTL genotype is estimated from the genotypes of the flanking marker loci and their distance from the QTL. Since there is usually uncertainty in the QTL genotype, the analysis is based on a sum of terms (one for each possible genotype, weighted by the probability of that genotype). The analysis point that yields the most significant association may be taken as the location of a putative QTL. The third method is called composite interval mapping (CIM: Zeng 1993; Jiang and Zeng 1995) or multiple QTL mapping (MQM: Jansen 1993) . Like simple interval mapping, this method evaluates the possibility of a target QTL at multiple analysis points across each interlocus interval. However, at each point it also includes in the analysis the effect of one or more markers elsewhere in the genome. These markers, sometimes called background markers, have previously been shown to be associated with the trait and therefore are each presumably close to another QTL (a background QTL) affecting the trait. The background marker genotypes are used in place of the (unknown) background QTL genotypes even though there will usually be differences due to recombination between these markers and the QTL. The inclusion of a background marker in the analysis helps in one of two ways, depending on whether the background marker and the target interval are linked. If they are not linked, inclusion of the background marker makes the analysis more sensitive to the presence of a QTL in the target interval. If they are linked, inclusion of the background marker may help separate the target QTL from linked QTLs on the far side of the background marker (Zeng 1993, 1994) . Each of the above methods differs in the form of the equation which is assumed to relate a trait value to the genotypes of marker loci. For single-locus mapping in a backcross population, the equation has the form
where Yi is the trait value for the ith individual, xi is the coded genotype of that individual, ei is the environmental effect in that individual, and a and b are parameters to be estimated. For SIM, the second term (bxi) is replaced by a sum of terms for each possible QTL genotype weighted by its probability. For CIM, the equation has this same sum and has, in addition, a bxi term for each background locus. For an intercross population, each bxi term may be replaced by a sum of two terms, one representing an additive component and one representing a dominance component. For example, the equation for CIM in an intercross population might be
where ba and bd represent the additive and dominance contribution of the target QTL and each bj represents the contribution of a background marker (assumed to be additive). For all methods, each mouse or RI strain provides the data for one equation, and the statistical problem is to estimate a and b (or each of several bs). Two methods have been used for this problem: (1) least-squares regression and (2) maximum-likelihood estimation using the estimation-maximization algorithm. Regression is computationally simpler, but it makes assumptions about the distribution of trait values that are only approximately true. Nevertheless, in practice, regression seems effective (Haley and Knott 1992; Martinez and Curnow 1992; Jansen 1993) . To avoid one of the assumptions about trait value distributions, it is possible to use a nonparametric statistic to evaluate the significance of an association, a measure based on the rank of each trait value rather than the trait value itself (Kruglyak and Lander 1995) . Recently, Whittaker, Thompson, and Visscher (1996) published a regression method which, for additive QTLs, is equivalent to the methods of Haley and Knott (1992) and Martinez and Curnow (1992) but which is computationally much simpler. This method allows the estimation of QTL position and effect from flanking-marker regression coefficients without fitting a regression equation at multiple points between the flanking markers. This increased speed will be especially welcome in the calculation of significance thresholds by permutation tests, described below. We do not want NTB clients to have to wait too long for results to be displayed. A constant problem in QTL mapping is establishing appropriate significance thresholds. First, although a likelihood ratio statistic can be calculated and interpreted as a c2 statistic, this ratio is only approximately c2 because the number of progeny is finite (and sometimes small). Second, because mapping involves multiple tests across the genome, significance thresholds must be correspondingly more stringent (Lander and Kruglyak 1995) . Permutation tests have been described which address both problems by calculating empirical thresholds specific for a data set and an appropriate range of the genome (Churchill and Doerge 1994; Doerge and Churchill 1996) . These empirical thresholds are essential, and the NTB will support their calculation. However, the calculations are time-consuming, and a recently proposed method offers a comparable empirical threshold with greatly reduced calculation time (Piepho, personal communication; Davies 1977, 1987) , so the NTB will offer this method as well. |
|||
|
Next Topic |
|||
|
|
|||
|
|
|||