![]()
Note to the reader: This is a revised edition of a paper published in Brain Research (1992;67: 285–91). The definitive original print version is available from Elsevier Press.
New figures, text, and links have been incorporated into the revision. Revised HTML (http://www.nervenet.org/netpapers/Rosen/MGBirth96/MGBirth.html) copyright ©1999 by Glenn D. Rosen
BIRTHDATES OF NEURONS IN INDUCED MICROGYRIA
Glenn D. Rosen, Gordon F. Sherman, & Albert M. Galaburda
Dyslexia Research Laboratory, Beth Israel Deaconess Medical Center; Division of Behavioral Neurology , Beth Israel Deaconess Medical Center, 330 Brookline Avenue, Boston MA 02215; Harvard Medical School, Boston, MA 02115.
All correspondence should be addressed to:
Glenn Rosen, Ph.D.
Department of Neurology
Beth Israel Deaconess Medical Center
330 Brookline Ave
Boston, MA 02215
Email:grosen@caregroup.harvard.edu
Freezing injury to the cortical plate of the newborn rat results in the formation of a focal region of cerebrocortical microdysgenesis resembling, in many ways, human 4-layered microgyria. Previous research has shown that neurons born during embryonic day (E) 20 migrate through the initial damage and take their place in the cell-dense layer of the microgyric lesion. The current study was conducted to determine (1) whether neurons generated earlier in development would be found in microgyric cortex and (2) whether the freezing injury would stimulate production of neurons postnatally.
Rat pups from mothers who were injected with S-phase markers on E15, E17, E19, and E21 were subjected to freezing injury of the cortex to induce microgyria on postnatal day (P) 1. Other pups received a freezing lesion and then pulse or cumulative injections of S-phase markers for the next 72 hours. Neurons born on E17 and E19 were found scattered throughout the cell-dense layer of the microgyric cortex. Early (E15) generated neurons were nearly absent in the microgyric cortex, and was there no evidence of postnatal induction of cortical neurogenesis. These results are considered in light of recent work demonstrating postnatal neocortical neurogenesis in response to early neocortical injury.
Freezing injury to the cortical plate of the newborn rat results in the formation of a focal region of cerebrocortical microdysgenesis resembling, in many ways, human 4-layered microgyria (1-6). Previous research has described various aspects of cytoarchitectonic disorganization that follow the freezing injury (3, 4). In brief, the destruction of neurons and glia induced by the freezing probe extends through the cortical plate, which at this age contains neurons normally destined to form layers IV-VIa. Occasionally the damage reaches through the subplate (VIb), but the pial membrane generally remains undamaged and radial glial cells, while damaged, are not eliminated. Reactive astrocytes and macrophages arrive in the damaged area within 24 hours of the injury, and repair of the damaged tissue peaks within the first week. Damaged radial glial fibers regrow, and supragranular neurons migrate through this damaged area, also within the first week. The newly formed supragranular layer overlies the cell-free area, and the damaged cortex begins to assume its adult-like microgyric appearance from postnatal day (P) 5 to P10. By P10 and lasting into adulthood, the microgyria is characterized by four layers comprising (i) an infolded molecular layer, (ii) a superficial neuronal layer (corresponding to ordinary layers II, III but less layered), (iii) a neuron-free zone (lamina dissecans), (iv) a deep neuronal layer (corresponding to ordinary layer VIb; Fig 1).
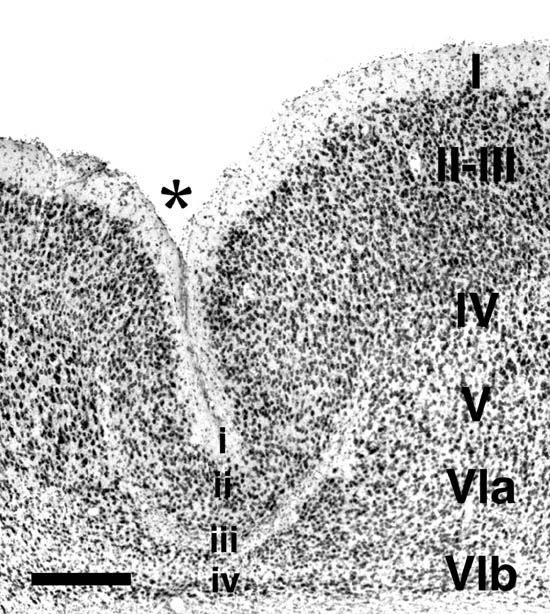 |
Figure 1. Photomicrograph indicating typical microgyria in the adult neocortex following focal freezing injury at P0. Asterisk indicates microsulcus. In comparison to the adjacent, undamaged six layered cortex (right), the typical microgyric cortex contains four layers (i-iv). Bar = 200 µm. |
What is not yet known, however, are the birthdates of neurons comprising the second microgyric layer (layer ii). In previous studies, animals with freezing lesions, the mothers of which had been injected with [3H]Thymidine on embryonic day (E) 20, showed labeled neurons only in the upper portions of the new layer ii (2, 3). These late-generated neurons were seen to migrate through the area of damage as well as through the area surrounding it. What was not known from these studies was the role, if any, of earlier or later generated neurons in the formation of microgyria following freezing injury. Kolb and colleagues have shown that rats who receive suction lesions of the frontal cortex at P10 have remarkable recovery of function and that neurons repopulate the lesioned area (7-10). They have further demonstrated that at least some of the neurons in the “repaired” frontal cortex were born after the lesion — a full 12 days after the end of normal cortical neurogenesis (11). We have shown radial glial-like immunoreactive fibers in the area of microgyria in P99 rats (a time well past the normal transformation of radial glial cells into astrocytes), and have speculated that the maintenance of this glial cell type may be related to the migration of neurons into the microgyria (12) and that at least some of these neurons may be born after the freezing injury.
In order to determine the range of birthdates of neurons in microgyric cortex, we injected pregnant Wistar rats with a single dose of an S-phase marker on either E15 (to label layer V-VI neurons), E17 (to label layer III-IV neurons), E19 (to label layer II-III neurons). In addition, we injected pregnant mothers after the end of neurogenesis (E21). The pups from these mothers were subjected to freezing injury at P1, and pups not previously injected with S-phase markers were given single or multiple injections of these markers from P1-P4 to determine if neurogenesis reinitiated after the freezing injury. The location of labeled neurons in the microgyria and in undamaged cortex was determined at P42.
MATERIALS AND METHODS
Protocol
Prenatal Injections. Six time-mated (the day that sperm was found in the vaginal smear was determined to be E1) Wistar rats (Charles River Laboratories) were injected intraperitoneally with [3H]Thymidine (3H-Td; New England Nuclear; 50-80 Ci/mmol; 5 µCi/g body weight) on either E15, E17, or E19. An additional 7 time-mated Wistar rats were injected intraperitoneally with 5-bromo-2'-deoxy-uridine (BrdU; Sigma) in saline with 0.007 NaOH (50 µg/g body weight) on either E15, E17, E19, or E21. On the day of birth (P0) or P1, the pups from these animals received either a freezing lesion or sham surgery. The pups were then sacrificed at P3 , P10, or P42 by intracardial perfusion with appropriate fixative (see below) and processed for serial sectioning in the coronal plane as described below. The 3H-Td-injected animals were processed for autoradiography and the BrdU-injected brains were processed immunohistochemically. The sections were examined for the presence of a microgyrus and the location of labeled neurons within and outside the microgyrus.
Postnatal Injections. Three time-mated Wistar rats were obtained and their pups randomly assigned to receive either a freezing lesion or sham surgery on P1. One litter was assigned to receive an injection of 3H-Td immediately (New England Nuclear; 50-80 Ci/mmol; 5 µCi/g body weight) and additional injections every 12 h subsequently until P4 for a total of 8 injections. Animals in the second litter received a single injection of 3H-Td at either 0, 24, 48, or 72 h after the freezing injury. The last litter was treated identically to the previous litter except that BrdU was injected (50 µg/g body weight). The pups were sacrificed at P10 or in adulthood (>P100), intracardially perfused with appropriate fixative, and processed and analyzed as described below.
Freezing Lesions. Focal necrotic lesions were induced based on a modification of the technique employed by Dvorák and colleagues (1, 2), and reported in detail elsewhere (4, 5). Briefly, pups were anesthetized via induction of hypothermia, and a small incision was made in the antero-posterior plane of the skin over the left cerebral hemisphere, exposing the skull. A cooled (-70°C) 2 mm diameter stainless steel probe was placed on the skull of lesion subjects, approximately midway between bregma and lambda, for 5 seconds. Sham subjects were prepared as above, except that the probe was maintained at room temperature. After placement of the probe the skin was quickly sutured, subjects were uniquely marked with ink injections to the footpads, warmed under a lamp and returned to the mother.
3H-Td Histology and Autoradiography. 3H-Td-injected animals were processed identically to those in a previous study (13). Briefly, the animals were deeply anesthetized with Pentobarbital (60 mg/kg i.p.) and were perfused transcardially with 0.9% saline followed by 4% paraformaldehyde (pH 7.4). The brains were removed from the skulls, postfixed for 24 hours, and then embedded in paraffin. The brains were cut coronally on a rotary microtome at 10 µm, and four series of every 20th section and four addition series of every 100th section for autoradiography trials were mounted onto glass slides. Mounted sections were deparafinized, hydrated and allowed to dry overnight. When dry, they were placed in autoradiographic slide holders (five per slide). Multiple slides from an animal’s brain were placed in the same holder. The slides were dipped in emulsion (Kodak NTB-2), placed in a light-tight container for 4-8 h to dry, and stored in a light-tight slidebox for 6-8 weeks at 0°C before developing (trial sections were removed periodically to test for progress). The slides were developed in Kodak D-19, counterstained with 0.05% thionin, dehydrated, cleared, and coverslipped with Permount.
BrdU Histology and Immunohistochemistry. The processing of the BrdU-injected animals was modified from Miller and Nowakowski (14). The animals were deeply anesthetized with Pentobarbital (60 mg/kg i.p.) and were perfused transcardially with 0.9% saline followed by a Carnoy’s true fixative (60% ethanol, 30% chloroform, 10% glacial acetic acid). The brains were removed from skulls, placed into 4% paraformaldehyde for 6-8 h and stored in 10% sucrose buffer overnight. The brains were then placed in 30% sucrose buffer until they sunk and sectioned coronally at 30µm on freezing microtome.
A series of every 5th section was processed for visualization of BrdU. Free-floating sections were washed twice in 0.1 M PBS for 5 min each. The tissue was then placed in 0.04% Pepsin in 0.1N HCl for 8-12 min and rinsed in PBS twice for 5 min each. The sections were immersed in 2N HCl for 20 min before being washed in PBS for 30 sec. The tissue was incubated overnight in the primary antibody (Becton-Dickinson) at a dilution of 1/200 at 4°C. After two rinses in PBS, the tissue was incubated with 0.45% biotinylated secondary antibody (Vector) for one hour and rinsed again in PBS before a 30 min incubation in 1.8% avidin-bound peroxidase complex. The tissue was rinsed twice in PBS and then twice in 50 mM Tris buffer (pH 7.6) and developed using 0.05% diaminobenzidine and 0.01% hydrogen peroxide diluted in Tris. After rinsing with Tris, sections were mounted on chrome-alum coated slides, dehydrated, counterstained with Methyl Green/Alcian Blue, coverslipped with Permount.
Analysis
The slides were examined under the light microscope and the presence of microgyria and other types of brain abnormalities recorded. The distribution of BrdU- and 3H-Td-labeled neurons was noted in the microgyria and comparisons were made with the distribution of labeled neurons in homologous regions of the opposite hemisphere as well as unaffected regions adjacent to the microgyria.
For postnatally injected animals, the microgyric and nonmicrogyric cortex was systematically examined using contiguous counting quadrilaterals (see 15 for details) spanning the entire length of the microgyria and the numbers of labeled and unlabeled cells counted.
RESULTS
Histologic appearance of lesion
Freezing lesions were consistent with those previously reported and depicted in Fig. 1 (1-5, 12, 16) and were located in parietal cortex. Depending on the plane of section, the microgyrus sometimes appeared as a wartlike excrescence on the cortical surface (see Fig. 2A). In some cases, the freezing injury led to two diverging microsulci that produced a true microgyrus sandwiched between the microsulci (see Fig 4A). The most common type of freezing injury recreated a microgyric four-layer cortex comprising (i) an infolded molecular layer, (ii) a superficial neuronal layer, (iii) a lamina dissecans, (iv) a deep neuronal layer (Figs. 1, 3A).
Location of labeled neurons
Prenatal Injections
Examination of the tissue from all ages of sacrifice yielded identical information. For clarity, the results from the animals sacrificed at P42 are illustrated here. In addition, there was no difference in results between the two S-phase markers and their results are pooled.
E15-injected. In the regions of the cortex unaffected by the freezing lesion, neurons labeled at E15 were located, as expected, predominantly in layers IV, V, and VI with the greatest concentrations being in layer V. In contrast, there were few heavily labeled E15-injected neurons in the microgyric regions (Fig, 2). When labeled neurons were seen in the region of the microgyria, they were located most densely in the layer subjacent to the lamina dissecans.
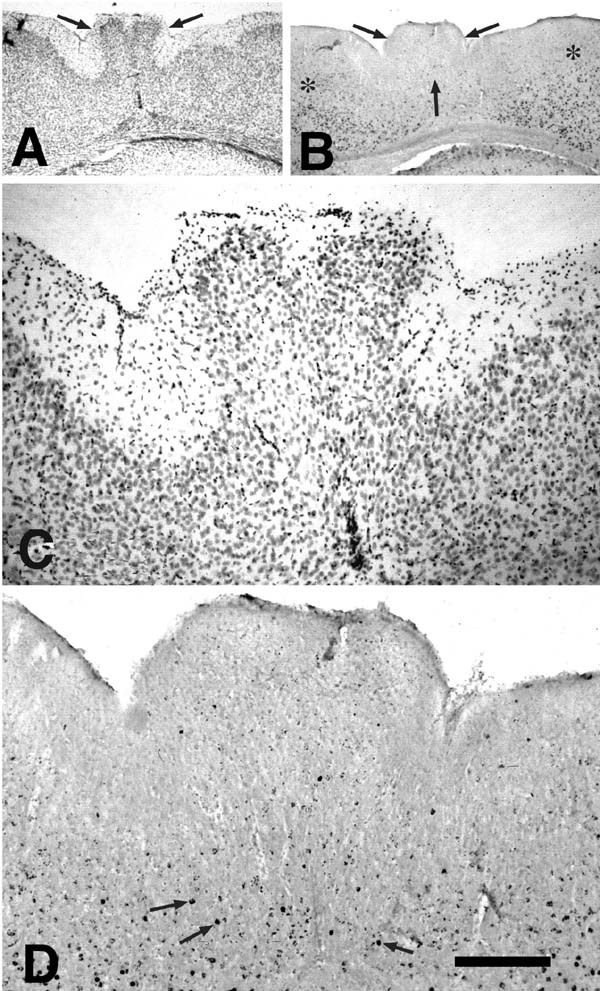
Figure 2. Rat injected with BrdU at E15. A. Nissl stained section of microgyric cortex showing wartlike excrescence (arrows). B. Section adjacent to A stained for BrdU. Note paucity of labeled neurons in the damaged area (arrows) in contrast to the higher concentration of these neurons in adjacent, undamaged cortex (asterisks). C. Higher power magnification of A illustrating the induced malformation. D. Section adjacent to C stained for BrdU demonstrating the paucity of neurons in the anomalous formation. Arrows indicate sample of labeled cells. Bar = 800 µm for panels A and B. Bar = 200 µm for panels C and D.
E17-injected. In the unaffected regions of cortex, neurons labeled at E17 were located, as expected, predominantly in layers III and IV, and relatively evenly scattered between the two layers. In the microgyric area, labeled neurons were seen in fairly high concentration in layer ii. The neurons were evenly dispersed throughout this layer, with no concentration either close to layer i or to the lamina dissecans (Fig 3).

Figure 3. Rat injected with BrdU at E17. A. Nissl stained section of typical microgyric lesion with microsulcus (asterisk). Arrows point to layer III-IV border of the undamaged adjacent cortex. Arrowheads denote lamina dissecans. B. Section adjacent to A stained for BrdU. In the adjacent, undamaged cortex there is a high concentration of labeled cells along the layer III- IV border and in the layers superior to it. Note the high concentration of neurons located in the microgyrus. Arrows for alignment with A. C. Higher power magnification of B illustrating the dense collections of labeled neurons scattered throughout layer ii of the microgyrus. ld = lamina dissecans. Bar = 800 µm for panels A and B. Bar = 200 µm for panel C.
E19-injected. Labeled neurons are seen mostly in layers II and III of the unaffected cortex and, like E17-labeled neurons, scattered throughout layer ii of the microgyric cortex (Fig. 4).
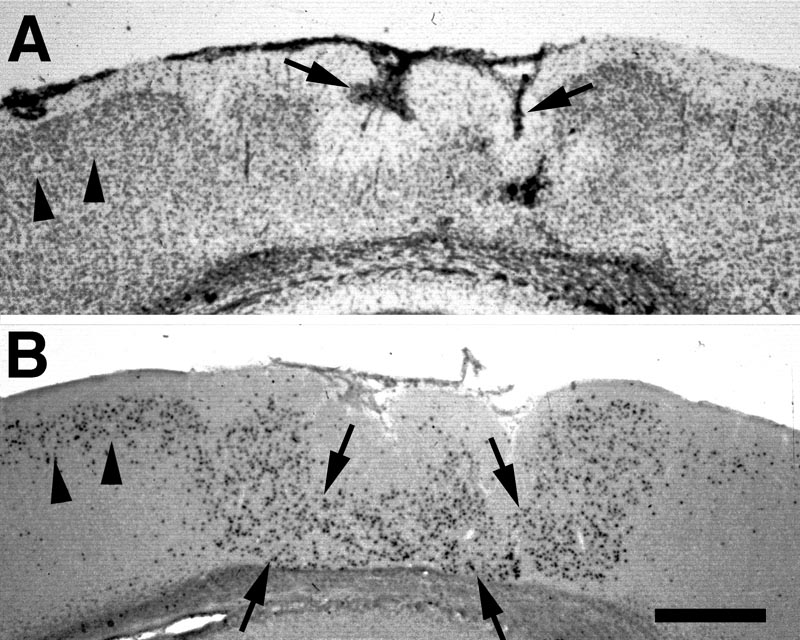
Figure 4. Rat injected with BrdU at E19. A. Nissl stained section of microgyric lesion with two microsulci (arrows). Layer II-III are denoted with arrowheads. B. Section adjacent to A stained for BrdU. In the adjacent, undamaged cortex there is a high concentration of labeled cells in layers II-III (arrowheads). Note the high concentration of neurons scattered throughout the microgyrus (arrows). Bar = 800 µm.
E21-injected. There were no labeled neurons in the affected or unaffected cortex following E21 injection of S-phase markers. This is in agreement with previous observations (13).
Postnatal Injections
Over 25,000 neurons were examined from pups representing each of the three litters and there was not a single instance of an unambiguously labeled neuron either in the microgyria or in undamaged cortex. In contrast, glia were heavily labeled throughout the neocortex (not illustrated).
DISCUSSION
Cortical Neurogenesis and Microgyria
It has previously been shown that neurons generated late in gestation (E20) are located in the uppermost regions of the second layer (2, 3) of microgyria produced by focal freezing injury to the cortical plate. In the current study, we too have found that neurons of the second microgyric layer are generated later in gestation. In addition, microgyric layer ii contains E17 neurons normally destined for layer III-IV of the undamaged cortex. Moreover, we found no lamination of E17 and E19 neurons in layer ii of the microgyria, with neurons from both birthdates being scattered throughout this layer. This lends further support for the designation of layer ii of the microgyria as a single lamina, despite its contiguity with adjacent undamaged neocortical layers II and III. Finally, we saw no evidence of neurons normally destined for the infragranular layers (born on E15) in the microgyria.
Following their last mitosis, neurons take 3 or 4 days on the average to reach their positions in the cortical plate (17). Thus, at the time of the freezing injury the only neurons present in the cortical plate are from the infragranular layers and probably some destined for layer IV. The freezing probe destroys these neurons in a cone shaped area underlying it, with occasional sparing of the subplate cells (3, 4). In subsequent days, later generated neurons, which were still in the germinal intermediate zone at the time of the lesion, migrate through the area of damage and form layer ii of the microgyria. Thus, the microgyria contains neurons generated both in the middle and later periods of corticogenesis. The mechanism for the migration of these neurons is not known, however. In the region of microgyria, radial glial fibers regrow into the area of damage (4) and maintain their typical long morphology well past the time that they would normally transform into astrocytes (4, 12). These non-transforming radial glial cells may act to enable the late migrating neurons to reach their final destination.
Alternatively, the late-generated neurons in layer ii may arrive from adjacent regions by collapsing into the area of necrosis. But the absence of early-generated neurons in layer ii argues against this hypothesis. It could be, however, that the reason E17 and E19 neurons, but not E15 neurons, arrive in the microgyria from adjacent areas is because by the time the region is reorganized (P5), the infragranular neurons have already established substantial local connectivity thereby precluding their move to adjacent areas. Later-generated neurons, on the other hand, are more recently arrived in the cortical plate and are therefore more malleable to movement.
In order to distinguish among these possibilities, the migration of labeled neurons at short survival times must be examined to determine whether these neurons migrate through the damaged area. Late generated neurons do indeed migrate through the necrotic area (1), suggesting that at least some portion of the neurons comprising layer ii of the microgyria do not arrive from collapsed adjacent regions.
Postnatal neocortical neurogenesis in the rat
As discussed above, Kolb et al. found that a P10 suction lesion of the frontal cortex induced cortical neurogenesis (11) which may explain the sparring of behavioral function which follows these lesions (7-10). Specifically, the neurons that repopulate the damaged region were comprised, at least in part, of neurons born after the suction lesion had occurred. We have found no evidence for induction of neocortical neurogenesis following freezing injury at P1. Thus, fibers with radial glia-like immunoreactivity that are seen in the microgyria into adulthood (12) are not maintained to aid the migration of postnatally generated neurons.
The difference between the current results and those of Kolb et al. could be due to the difference in location between our microgyria and the suction lesions (11). None of our microgyria were located in the frontal cortex, and the location of our microgyria (parietal cortex) was in the identical region where Kolb et al. found that neonatal lesions did not result in functional recovery (18). Another possibility is that we did not extend the injection of S-phase markers long enough into the post-natal period. This is unlikely because the lesioned region begins to take on its microgyric appearance around P5 (4), and the neurons comprising the microgyria could not have been born after P4. Further, the postnatally generated neurons in the Kolb et al. study were born within 48 h of the lesion (11). Thus, if neurogenesis is initiated as a result of developmental injury, one would expect that it would occur shortly after the injury occurred. We cannot exclude the possibility that neurons generated later than P4 could form at least part of the microgyria. It should also be noted that timing of the lesion itself may play an important role. For example, suction lesions prior at P5 do not result in the same type of filling in of the lesion cavity (Kolb, personal communication) as seen following lesions after P7. It is therefore possible that the type of injury necessary to form microgyria occurs too early to induce neocortical neurogenesis.
CONCLUSIONS
We have found that the cell dense portions of induced microgyria are formed by neurons generated between E17 and the end of neocortical neurogenesis. In unaffected cortex these neurons comprise layer IV and supragranular layers. In contrast, we found no evidence of large numbers of early generated neurons in the microgyria. Based on these results, we confirm previously hypothesized scenarios for the formation of microgyria: Following freezing injury at P0 or P1, the early-generated neurons comprising the cortical plate at the time of the insult are killed. Later-generated neurons migrate through the area of cortical necrosis and take their place in the reorganized cortex in the unlaminated layer ii of the microgyria. Because of the absence of early-generated neurons in layer ii of the microgyria, it is unlikely that neurons from adjacent undamaged cortex collapse into the necrotic area.
ACKNOWLEDGEMENTS
The authors wish to acknowledge the technical assistance of Judy Richman, Douglas Press, and Lisa Stone Garcia. This work was supported, in part, by HD20806.