 |
| |||||
|
| ||||||||
|
| ||||||||
Home  Publications Publications |
|
|
Note to the Reader This is a preprint of a paper now published in Behavior Genetics (Vol 31, issue 1, 2001). Please cite this work as: Belknap JK, Hitzemann R, Crabbe JK, Phillips TJ, Buck KJ, Williams RW (2001) QTL analysis and genome-wide mutagenesis in mice: Complementary genetic approaches to the dissection of complex traits. Behav Genet 31:5–15. The definitive print and html versions are available on the Behavior Genetics web site. Copyright: Kluwer/Plenum.
Print Friendly
Abstract Quantitative genetics and QTL mapping have undergone a revolution in the
last decade. Progress in the next decade promises to be at least as rapid,
and strategies for fine mapping QTLs and identifying underlying genes will
be radically revised. In this commentary we address several key issues:
first, we revisit a perennial challenge–how to identify individual genes and
allelic variants underlying QTLs. We compare current practice and procedures
in QTL analysis with novel methods and resources that are just now being
introduced. We argue that there is no one standard of proof for showing QTL=gene;
rather, evidence from several sources must be carefully assembled until
there is only one reasonable conclusion. Second, we compare QTL analysis
with whole genome mutagenesis in mice and point out some of the strengths
and weakness of both of these phenotype-driven methods. Finally, we explore
the advantages and disadvantages of naturally occurring vs mutagen-induced
polymorphisms. We argue that these two complementary genetic methods have
much to offer in efforts to highlight genes and pathways most likely to
influence the susceptibility and progression of common diseases in human
populations. QTLs that are critical in behavior and neurological disease are being
mapped at a rapid pace in mouse and human populations (Belknap et al., 1997;
Crabbe et al., 1999; Burmeister, 2000; Tecott and Wehner, 2001; Phillips et
al., in preparation). Numbers of significant QTLs for behavioral and CNS
traits in mice have increased more than tenfold in the last 5 years–from 3
in 1995 to over 40 by 2000. This issue of Behavior Genetics covers a
cross-section of this exciting work. We hope to shed light on two questions
that are important in such a rapidly evolving field: where do we want to be
in the next few years, and how can we get there? This involves three key
issues. One issue concerns the methods and prospects for identification of
single genes associated with well mapped QTLs. The second issue is the
relative merits of QTL versus whole genome mutagenesis approaches. Finally,
we discuss the pros and cons of genetic variation induced by mutagens
compared to that found in existing mouse strains and lines. We explore these
issues in the context of looking at the problems that beset both QTL and
genome-wide mutagenesis screens of complex traits. A provocative essay in a recent issue of Nature Genetics reviewed
the obvious challenges associated with gene identification (Nadeau and
Frankel, 2000) and made the case that mutagenesis of the entire genome
provides an alternative, more rapid, and more certain route around an
apparent "roadblock" in QTL analysis. We agree with facets of this review,
but we disagree with the gloomy assessment of the current status and
immediate prospects of QTL gene identification. In this commentary, we
revisit this issue and present QTL analysis and mutagenesis as complementary
(rather than competing) methods that will both need to be used to screen an
entire genome for subsets of genes that influence specific traits. These two
genetic strategies share numerous scientific and clinical objectives (Lander
and Schork, 1994; Takahashi 1994), but there are key differences between
methods, realms of application, scientific goals, and the areas of expertise
of practitioners who work in these fields. We also consider new
opportunities associated with the influx of sequence data, comprehensive CNS
gene expression data, and high-resolution mapping resources. Finally, we
note some of the problems we see in whole-genome ENU mutagenesis screens as
applied to genetically complex traits such as behavior. Our conclusion is
that QTL analysis will be a lively and crucial partner in functional
genomics in the foreseeable future as will genome-wide mutagenesis screens.
Without losing sight of genuine challenges, even pessimists should be
encouraged by the continued hybridization of quantitative and molecular
genetics. Standard operating procedures: QTL analysis and genome-wide mutagenesis
in mice A complex trait is a phenotype resulting from multilocus determination
coupled with multiple environmental influences. The first step in QTL
analysis is to select a cluster of closely associated complex traits that
reflect a single well-defined biological problem. Biological questions and
specific phenotypes drive the entire program. The operational difficulties
of measuring important traits usually do not dissuade investigators, as
shown by the employment of elaborate behavioral paradigms,
immunohistochemical procedures, and quantitative electron microscopy in the
last few years to map QTLs. Investigators are often specialists with highly
focused interests in the genetic basis of the very particular traits under
study. They also usually share strengths in statistical and quantitative
genetic analysis. QTL studies are usually carried out in a single laboratory
or as part of small group projects. In a typical QTL analysis of a complex trait, a mapping population is
generated by crossing two highly differentiated progenitor strains or lines
of mice. Most often, several hundred F2 or backcross progeny are tested and
genotyped genome-wide. Sometimes, recombinant inbred strains are used as a
preliminary screen coupled with other mapping populations. The initial goal
is to dissect existing continuous or quantitative genetic variation into its
component loci (QTLs), and to map them to broad chromosomal regions. This is
only the first step; the aim of this effort is to rapidly return to the
biology to gain a better understanding of underlying molecular and cellular
control of the target trait as well as at the systems and organismic
levels–initially in mice, but ultimately, in human populations (Williams,
2000). The most effective route to this goal is to identify genes
unambiguously associated with QTLs. These genes then become key entry points
from which to explore the network of genes, proteins, pathways, systems and
environments important in the determination of a complex trait. Strong
homologies between genes in mice and humans usually guarantee overlapping or
even near identical biological function. Furthermore, polymorphic genes in
mice may also be polymorphic in humans; at least functionally important
allelic variation may be discovered in the same pathways if not the same
genes. The first step in a typical genome-wide mutagenesis screen is to select a
set of traits that can be scored rapidly and with high throughput in a
thousand or more mice. The choice of traits is governed as much by economy
and throughput as by biological interest. For this reason, directly and
readily observable abnormalities, such as dysmorphology or abnormal motor
movements, figure prominently in the range of traits chosen for study.
Investigators who lead this type of research are usually molecular
geneticists with special competence in gene structure and cloning (See
reviews by Brown and Nolan, 1998; Noveroske et al., 2000; Nolan et al.,
1997, 2000; Hrabe de Angelis et al., 2000; Schimenti and Bucan, 1998; Wells
and Brown, 2000). ENU screens of adult mice are massive and very expensive
undertakings that are now often coordinated at a national level (e.g. Hrabe
de Angelis et al., 2000; Nolan et al., 2000). In a typical whole genome mutagenesis screen, inbred male mice are
treated with a strong alkylating agent, ethylnitrosourea (ENU), to induce
several hundred germline mutations per mouse. They are crossed to wild type
females of the same strain, and large cohorts of offspring are run through a
gauntlet of tests to identify the individual outlier or extreme-scoring mice
most likely to bear a large-effect Mendelian mutation. These outlier mice,
usually defined as those that are >3 SD units from the mean, are each
progeny-tested to determine whether their abnormality segregates bimodally
in their offspring with the expected 1:1 or 3:1 Mendelian ratios. Those that
pass this test (a minority of all extreme-scoring mice) are subsequently
mapped at moderate resolution (10-20 cM) in a cross to a different inbred
strain using genome-wide markers and methods similar to QTL mapping studies.
To fine-map each mutation to a 1 cM interval (95% confidence interval)
typically requires a cross of about 500 progeny (Wells and Brown, 2000). At
this level of precision, brute-force sequencing can be effective in
identifying the ENU-induced mutation since there are expected to be about
750 mutations throughout the genome, or about one every 2 cM (Schimenti and
Bucan, 1998). These induced mutations, because of their large-effect
Mendelian patterns of inheritance and fewer polymorphisms to sort through,
currently have an important advantage over QTLs in ease of gene
identification. Isolating Genes underlying QTLs What are the prospects for gene identification for QTLs? At present, it
is difficult to establish connections between continuous phenotypic variants
and the associated set of mapped gene variants (Darvasi, 1998). Nadeau and
Frankel describe this task as a "long and bumpy road." However, it is worth
pointing out that QTL studies that were almost inconceivable a decade ago
are now routine. As we discuss below, many new developments are on the
horizon that will fundamentally alter how we identify genes underlying QTLs.
To modify the metaphor, that "long and bumpy road" is better seen as a
high-speed highway under construction. Current and evolving developments will greatly simplify fine mapping and
gene identification. One key factor is, of course, complete genome sequences
for the most commonly-used inbred progenitor strains. Provisional coverage
is already available for 129/SvJ, DBA/2J, and A/J from Celera Genomics (see
www.pecorporation.com/press/prccorp101200.html), and C57BL/6J is now being
sequenced as part of an NIH-supported effort (see www.ncbi.nlm.nih.gov/genome/seq/Mmprogress.shtml).
As a result, generating comprehensive lists of functional polymorphisms
between these four major strains will soon be routine for any part of the
genome. Among crosses between any pair of these strains, the source of all
QTLs will be known. A parallel situation will also exist at the mRNA and
protein levels; the development of array-based methods will reveal numerous
strain differences, the source of many QTLs. In a few years, QTL studies
will begin with complete lists of differences in gene expression and protein
levels in several commonly-used progenitor strains of mice. This is already beginning to happen. For example it is now possible to
generate a list of differences in the expression level of 7169 genes in the
hippocampus between strains C57BL/6J and 129 (Sandberg et al., 2000; ftp://ftp.gnf.org/pub/papers/brainstrain/).
Identifying hippocampal-dependent QTLs from crosses between these two
strains may be more like "cruising in a Cadillac" than a hard road trip. In
contrast to the enormous benefit of these developments for QTLs, the benefit
for ENU mutants will be less because much of this work at the sequence,
transcript and protein levels must be done anew with each newly-created
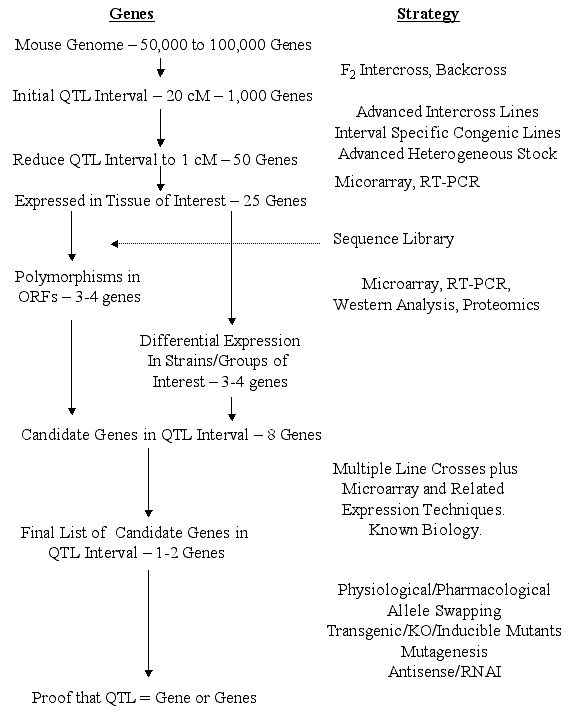
mutation of interest. Steps from QTL to gene. Surprisingly, the actual steps involved in
moving from QTLs to genes have received only cursory attention. The
following outline and flowchart (Figure 1) summarizes our views of the
likely steps. We assume that a QTL has been refined to intervals of 1 cM
(95% confidence interval) that will contain an average of about 50 genes.
[This is based on an estimated 75,000 genes distributed over 1450 cM, a
worst case scenario since recent estimates suggest only 30,000 to 40,000
genes.] The 1-cM criterion is not unreasonable since several behavioral QTLs
have now been mapped with high LOD scores and impressive precision (Crabbe
et al., 1999; Demarest et al., 2001; Talbot et al., 1998; Fehr et al., in
press). We assume that the cells and tissue types related to the phenotype
are known or strongly suspected. This will almost always be the case for
morphometric traits (Le Roy, 2001, Williams et al., 2001, both in this
issue), but for higher-order behavioral traits, inferences will be
provisional at early stages of analysis. Genes Expressed in the Tissue of Interest. Only a fraction of the
genes within the QTL interval will be expressed in the tissue of interest at
some point in the life of the mouse. Microarray technology can easily
address this problem for specific subsets of genes. At most, about half of
the genes in an interval will be expressed in brain (Sandberg et al., 2000),
and consequently, only this half need be considered for further testing for
most behavioral traits. Carrying out expression analysis at an early stage
is based on the assumption that the sequence information for multiple inbred
strains will be of variable accuracy for the next several years; thus, some
sequencing will still be necessary. Therefore, a reduction in the number of
candidate genes by expression studies should save time. However, once
accurate sequences are readily available, expression and proteomic studies
would more efficiently become part of the end game. Polymorphic Genes - Open Reading Frames (ORFs). From the completed
genomic sequence for the primary inbred strains of interest (e.g. C57BL/6J
and DBA/2J), it will be possible to screen and map the polymorphisms within
the ORFs "in silico". Nonconservative amino acid substitutions
between progenitor strains in ORFs are relatively uncommon (10-20%) (Buck
and Finn, 2000; Fehr et al., in press) and we assume that about 50% of these
polymorphisms will have functional significance. With this estimate, the
number of genes with functional polymorphisms in the ORFs is reduced to
about 3-5 genes per 1 cM that will merit serious consideration. In the
short term, the function of some (if not most) of these genes will not be
well known, but we can often make strong inferences from homology with other
better-characterized genes. Strategies for determining which (if any) of
these polymorphisms may be associated with the QTL are described below. Expression Array Approaches A useful approach would be to look for
expression differences among the strains and treatment groups defining the
phenotype; this could be combined with the methods discussed above,
being mindful that important differences in expression may have occurred
earlier in development. There are several problems with expression array
analysis that deserve comment. First, the method is only semiquantitative
and any apparently significant result will require confirmation with a
quantitative technique, e.g., quantitative RT-PCR. Second, differences in
RNA expression do not always lead to differences in protein content; thus,
evidence that protein levels have actually changed must be provided, e.g.,
quantitative Western analysis. Third, small but functionally relevant
differences in gene expression may not be detected. And fourth, not all
relevant genetic sequences are currently available for analysis.
Fortunately, these problems are likely to be relatively short-lived and
should disappear with advances in expression technology and proteomics. Emerging data indicate that only a small percentage of genes will show
detectable differences in expression between pairs of inbred strains.
Sandberg et al. (2000) found that about 1% of genes varied in expression
level by more than two-fold between the C57BL/6 and 129 strains across
several brain regions. Therefore, we would expect an average of less than
one gene showing detectable differential expression between a pair of inbred
strains within a 1 cM interval. While expression studies cannot by
themselves rule out candidates, they can certainly provide important support
for particular candidates. Narrowing the List of Potential Candidate Genes One could argue that
although thethe "gene list" for the QTL interval can be substantially
reduced based on the above considerations, to be efficient we need to reduce
the number to one or two candidates. In some cases, there will be an obvious
candidate that is plausibly associated with the phenotype, such as the
cluster of GABA-A subunit genes in the region of a QTL affecting both
alcohol and pentobarbital withdrawal severity (Buck et al., 1997, 1999; Buck
and Finn, 2000). To date, this opportunistic approach has led to successes
in moving from QTL to gene. However, in general we assume that the function
of the remaining candidates will either be unknown or only partially known.
Can the list of candidates be further reduced? One practical method is to
use much higher resolution community-based mapping resources capable of sub-cM
precision. For example, chromosome substitution strains (Nadeau et al.,
2000) can be rapidly converted into interval-specific congenic strains for
high resolution mapping (Darvasi, 1998; Williams, 1999). Heterogenous stock,
advanced intercrosses and recombinant progeny testing are additional
effective methods that can attain 0.5 cM precision or better (e.g., Talbot
et al., 1998; Mott et al., 2000; Darvasi, 1998; Lyons et al., 2000; Fehr et
al., in press; Demarest et al., this volume). Recombinant inbred strains
could be easily extended for high resolution mapping. A set of 100 BXD RI
strains would permanently archive about 6000 recombination events and this
would often be sufficient to confine a QTL to a 0.25 cM interval using RIST
(Darvasi, 1998) and other methods. Such precision mapping coupled with the
above considerations will often narrow the list of plausible candidates to
just one gene. The End Game - QTL to Gene. We believe that existing technology in
combination with technology that will soon be acquired will soon lead to one
or two very strong candidates without relying on knowledge of gene function.
The question that now arises is what will be acceptable as proof that a
particular gene underlies a QTL. Nadeau and Frankel (2000) claim that an
allele "swap" should be the formal proof of identity, or the "gold standard"
(our words). Certainly, if the allele swap produced the expected phenotypic
changes, this would be a powerful argument. However, in simpler systems such
as bacteria, allele swapping has not always produced the expected phenotypic
results because of genetic background (epistatic) effects (e.g., Malke et
al., 2000). Thus, it would appear that we need to consider additional means
of "proof." We propose the scheme depicted in Figure 1 as a reasonable
strategy for determining whether a gene is responsible for a phenotypic
difference. The following are some likely strategies. One, the process described above for reducing the number of candidates to
one can provide compelling evidence. Two, physiological/pharmacological approaches can be used for genes where
function is known, e.g. will specific inhibitors of the gene product produce
the expected phenotype? Three, transgenic (overexpressing, underexpressing, null) mutants should
be useful especially for genes of unknown function, despite the well known
limitations of this approach. Some of these limitations can be overcome
through tissue-specific inducible mutants (Tecott and Wehner, in press). Four, antisense and related techniques can be used to knockdown genes
transiently; this approach is particularly well suited to "brain"
phenotypes, where one may wish to knockdown gene function in only a specific
region or nucleus. Viral transfer strategies can be used to produce the
opposite effect--targeted gene overexpression. Overall, we would argue that there is no single proof of identity, or
"gold standard", for proving that a gene underlies a particular QTL. Rather,
proof will rely on the careful assembly of evidence from several sources
that leads to only one reasonable conclusion. Finally, it is important
to note that given the current and expected advances in genomics, proof that
QTL = gene can occur largely independently of any knowledge of gene
function, the most difficult scenario. However, in the shorter term,
knowledge of gene function vis a vis the phenotype will no doubt be
an important contributor to gene identification successes. Natural vs Mutagen-Induced Genetic Variation What advantages and disadvantages do we incur when we choose to induce
genetic variation rather than rely on naturally occurring variants? The
advantages are great. Rendering presently monomorphic loci into polymorphic
ones is a marvelous capability. This opens a whole range of genes for
investigation that cannot be studied by QTL or other nonmutagenic
approaches. The prospects are especially exciting for the study of genes
that direct early development since they normally cause little variation.
Working with loci that have large rather than small effects is another
important advantage. While naturally occurring or "spontaneous" mutations
have led to many valuable genetic disease models, the ones that have most
often led to gene identification have been principally large effect
mutations showing single locus or Mendelian inheritance. Recent studies with
neurological and development mutants such as vibrator (Hamilton et
al., 1997) are prime examples. ENU mutagenesis seeks to amplify on this
successful approach by systematically extending the range of large effect
mutations available for study. Largely for these reasons, two of us are
committed to carrying out mouse ENU studies in our laboratories. While we
support the growing interest in applying genome-wide mutagenesis to complex
traits, this approach should not be considered uncritically, especially for
behavioral traits. Therefore we ask--are there disadvantages to
mutagen-induced variation? Yes—and in the next several paragraphs we
describe seven drawbacks that should be considered in designing such
studies. Some of the drawbacks stem from errors in the process that must be used
to detect and recover valuable mutations. There are three steps involved in
this process, (1) the phenotypic screen to detect individual outlier mice,
(2) the progeny test to determine whether the outlier phenotype is due to
single locus inheritance, and (3) genome-wide chromosomal mapping to provide
a further check on single locus inheritance as well as to begin gene
identification efforts. For complex traits showing considerable background
variation, there are Type I (false positive) and II (false negative) errors
to consider at each of the three steps that rarely matter with simple traits
showing little background variation. We argue below that such errors can
greatly diminish the usefulness of genome-wide mutagenesis in the study of
complex traits, especially behaviors, which are often the most complex of
all. First, for some traits, it may be very difficult to identify which
animals bear a valuable mutation. The favorite example of a successful mouse
mutagenesis experiment is the isolation of the clock gene by
Takahashi and his group (Vitaterna et al., 1994). The success of this
experiment depended at least in part on the extremely small variability in
circadian rhythm photoperiod in the background inbred strain. Thus, a single
outlier mutant mouse could easily be detected against a nearly uniform
phenotypic background. For complex traits, the existence of many
environmental influences causing mice to vary phenotypically will make this
task much more difficult (Tarantino et al., 2000). This problem arises
because the detection of a valuable mutant hinges critically on the
phenotype of a single outlier mouse relative to the background
phenotypic variation. We expect that as environmental variation increases,
fewer outliers will be apparent against an increasingly variable phenotypic
background. This will have the effect of reducing the outlier rate, or
percent of outliers, and thus the yield of recovered mutations. Moreover,
the risk of false-positive outliers, i.e., those not due to a
large-effect single locus mutation, increases as the background variance
increases, making the recovery of valuable mutations more difficult. Increased environmental variance is often associated with reduced
reliability or repeatability of measurement (Falconer and MacKay, 1996).
This problem is evident when a mouse, appearing to be an outlier when first
tested in a phenotypic screen, may not be an outlier when tested a second
time on the same assay. This has been reported for some behaviors (Nolan et
al., 2000) and may reflect the regression toward the mean expected when
reliability is less than perfect (Falconer and MacKay, 1996). Retesting of
each mouse may be needed to insure that an outlier mouse truly is an
outlier, or in other words, to reduce false positive errors in the
phenotypic screen. For many behaviors, retesting is not feasible because
only the first test is valid (e.g., learning or anxiety), or because the
first test alters the outcome of later tests, so the phenotype is no longer
the same. False positive outliers at this stage are troublesome because they
lead to progeny testing with little hope of passing the progeny test. This
reduces the percent of all outliers ultimately shown to be valuable
mutations. The difficulty and cost of recovering each valuable mutation are
also increased. Similar problems arise at the progeny-testing step. To pass the progeny
test, the offspring phenotypes must be distributed bimodally as expected
from 1:1 or 3:1 Mendelian ratios of wild type to mutant genotypes. To
generate a bimodal rather than a unimodal distribution, a single mutant
locus would have to account for two-thirds or more of the phenotypic
variance (Belknap et al., 1993). Put in other words, the variance due to the
mutant locus would have to be at least double that of the background
variance for detection to occur. [Unfortunately, because the animals cannot
be genotyped to differentiate mutant from nonmutant genotypes, the trait
distribution is our only means for separating the two genotypes.] As the
background variance increases, the probability diminishes that a single
mutant locus will meet this criterion in a progeny test. This has the
undesirable effect of decreasing the percent of extreme-scoring mice passing
the progeny test by increasing the frequency of false-negative errors, which
are valuable mutants that are not detected. Alternatively, one could abandon
the bimodal distribution requirement and simply require that the progeny
score differently than the background strain by a less stringent criterion.
But this would increase the rate of false positives, which would then
undergo expensive chromosome mapping studies with little hope of recovering
a valuable mutant. Either way, the difficulty and cost of recovering each
valuable mutation will be substantially increased as a function of the
magnitude of the background variance. Second, the successful screening and mapping of a mutant is just the
beginning of the process of determining whether it has any utility to
increase our understanding of pathways important for a complex trait. Much
effort must often be expended to answer the basic question—what is this
mutant good for? Let’s assume in carrying out a screen for learning ability,
we find a mutant that exhibits almost no learning of a given task.
Considerable effort could be devoted to mapping and characterizing this
mutant, only to find that its performance is due to reasons unrelated to
learning--a sensory or effector deficit may be the cause. Another example is
a mutation that seriously impairs vision--it will likely be detected and
recovered on a screen for anxiety since most assays for this trait presume
normal vision (e.g., Cook et al., 2000). For lack of a better word, we call
these trivial (for a given trait) mutations because they are unlikely to
shed light on the fundamental processes involved in either learning or
anxiety. [Of course, a trivial mutation for one trait may serendipitously
prove to be valuable for another trait.] The more complex the trait genetically, the more genes (and pathways)
will be involved across several organ systems. Since many if not most of
these pathways will be trivial to an understanding of the trait, it can be
difficult to sort out which mutants are trivial and which are not. Because
trivial mutations will pass the progeny test as readily as nontrivial ones,
they will undergo expensive mapping efforts with little hope of being
particularly valuable. This implies that greater genetic complexity can be
expected to lead to a reduced recovery rate of nontrivial or valuable
mutations, as well as increase the cost and difficulty of recovering each
valuable mutation. [There are undoubtedly trivial QTLs as well, but this is
much less of a problem compared to induced mutants.] Third, mutagen treatment induces an average of several hundred
mutations in every mouse. For genetically very complex traits, defined as
those with large numbers (potentially many thousands) of mutable
trait-relevant genes, phenotypic screens will likely select individual
outlier mice with several trait-relevant mutations (polymutations),
not just one. If so, then the effects of the individual polymutations will
be much smaller than expected, thus compromising one of the advantages of
this method, the production of large-effect Mendelian mutations. The more
complex the trait genetically, the more often trait-relevant polymutations
will predominate among the outlier mice. These mice are unlikely to pass the
progeny test because each polymutation is unlikely to account for
two-thirds or more of the trait variance required for detection. To make
matters worse, the effects of the smaller polymutations will add to the
background variation, making a bimodal distribution due to the largest of
them even less likely. This has the effect of reducing the percent of
outlier mice passing the progeny test, and increasing the cost and
difficulty in recovering each mutant. We now have another reason for
believing that increased genetic complexity will be associated with a
diminished recovery rate of valuable mutants. Moreover, since mutant gene
mapping requires crosses between different inbred strains, large-effect
mutations are essential if they are to be discriminable from the QTLs also
segregating in the mapping population. One important implication of the last three points noted above is that
there may be subsets of complex traits, particularly behaviors, with outlier
rates and recovery rates so low that the mutagenesis approach is only of
marginal utility. In this situation, one could adopt a brute-force strategy
and progeny-test much larger numbers of mice to increase the probability of
recovering some nontrivial mutations. To do this implies that we must either
relax our standards for what qualifies as a positive result for the first
(outlier detection) or second (progeny testing) steps (which increases
false-positives and further reduces recovery rates), or we must expand the
total size and scale of the screen well beyond that needed for simple traits
such as kinked tails and circling movements. Either way, the cost and effort
will be greatly increased. For the reasons given above, recovery of each valuable mutant for a
complex trait is likely to be much more difficult and expensive than for a
simple trait. To be sure, complex traits offer more targets for mutagenesis
compared to simple traits, and thus the potential number of valuable mutants
is greater per trait, but the increased difficulty and cost in
recovering each mutation will take its toll on the usefulness of this
method. This conclusion runs counter to that implied by some proponents of
genome-wide mutagenesis (e.g., Nadeau and Frankel, 2000). Unfortunately, for
those of us interested in behavior, it does not appear that ENU is going to
save us from the complexity of our preferred phenotypes. At present, the
analysis of complex behavioral traits by induced mutagenesis is too new to
allow an adequate empirical test of our concerns, but preliminary results
thus far are consistent with our expectations (e.g., Sayah et al., 2000;
Nolan et al., 1997). Fourth, there are strong biases in favor of mutation detection in
some genes over others, and this reduces the proportion of all
trait-relevant genes likely to be recovered in mouse ENU screens. Mutated
genes most likely to be detected are those that have large effects on the
phenotype resulting from base-pair substitutions at any one of hundreds of
sites within the gene. A good example is a gene where point mutations at
many sites all lead to premature stop codons; thus, this gene will likely
emerge often in a screen while genes without this property may go
undetected. [Multiple detections of the same gene are already apparent in
ongoing mouse ENU studies, which can be useful if multiple allelic series
are created, but this does not help the detection bias problem.]
Trait-relevant genes unlikely to emerge are those with considerable
phenotypic effects, but not enough to induce an outlier mouse no matter
where the site of the mutation. For such genes, even null or constitutive
mutants won’t be enough to lead to their detection and recovery. This
detection bias will likely be greatest for genetically complex traits
compared to simpler ones because of the higher frequency of trait-relevant
genes whose mutated effects on the phenotype are too small to allow their
recovery. Thus, the claim that all trait-relevant genes are potentially
recoverable is highly questionable for complex traits. In addition, mutants
showing recessive inheritance (the majority) are much less efficiently
detected than those showing dominance, another major source of detection
bias. Strong bias also applies to the range of all behaviorally important
phenotypes amenable to study by genome-wide mutagenesis. Phenotypic screens
of mutagenized mice require large numbers of animals, much more than a
typical QTL study. This introduces a bias for practical reasons in favor of
traits that require little time or effort to phenotype each mouse, and do
not affect the outcome of subsequent tests of other phenotypes carried out
on the same mice. This is one reason why dysmorphological traits predominate
in major mouse ENU screens since they can be detected by simple observation,
and among behaviors, abnormal locomotor activity is a favorite phenotype. The mutant detection and recovery bias against traits with large amounts
of background variation has already been mentioned. Complex traits that show
floor or ceiling effects (common with behaviors) do not work well because of
the truncated distribution, making the identification of outliers almost
impossible; however, these traits often work well for QTL studies. Also,
traits that require sacrificing the animal to measure them, such as
neurochemical or neuroanatomical measures, can be studied easily by QTL
methods using replicated, isogenic genotypes inherent in recombinant inbred
strains, congenics, recombinant congenics or chromosome substitution strains
(consomics), but do not lend themselves well to mutagenesis screens for
outlier genotypes that are neither isogenic nor replicated when the
phenotypic screen is performed. This disadvantage means that outlier mice
must serve as breeders or as sperm or ova donors prior to sacrifice for
phenotyping. Therefore, detecting such outliers in the first place will
require sperm or ova freezing for all of the several thousand mice in
the screen prior to phenotyping, followed by in vitro
fertilization/implantation procedures to propagate the outlier genotypes.
These burdensome requirements make such traits undesirable if not unworkable
for mutagenesis screens. Fifth, phenotype-driven mutagenesis screens, for practical reasons,
are designed to detect only those mutations with the largest effects on a
given trait. These are precisely the ones most likely to cause developmental
compensation on a scale seen in some targeted mutagenesis (knockout) mice (Gerlai,
1996; Bilbo and Nelson, 2001; Crawley, 2000). Indeed, the majority of
recovered mutants from mouse ENU screens are null mutants (Neveroske et al.,
2000). This can introduce a troubling confound, for we will always be unsure
of the degree to which a particular phenotype we observe in a mutant strain
is due to an induced mutation or to other nonmutated genes whose
expression has changed to compensate for the mutation effect. In addition,
perturbation of pathways by a mutation may be so great as to cause a cascade
of secondary effects not seen in normal mice, many of them in pathways far
removed from the site of the mutation. In such cases, the question
becomes-—to what degree does a particular observed "phenotype" of a mutant
reflect aberrant secondary effects? Strong secondary effects, particularly
during development, can obscure the normal function of affected pathways and
greatly complicate inferences about the cause of an observed phenotype
because of their largely unknown nature and because there can be so many of
them. In addition, such effects often disrupt the normal interplay among
genes (epistasis), an increasingly important focus for complex trait
studies. Secondary effects pose other problems as well. Among those mutants that
are viable, many show reduced health and vigor that may nonspecifically
confound a phenotypic assay when a "sickly" mutant genotype is compared to a
more vigorous wild type (normal) genotype. When differences are found (e.g.,
the mutants may be less active), we may incorrectly attribute this to a
specific pathway of a known mutation when the true explanation lies with
unknown secondary effects serving to impair health or vigor. This problem is
likely to be of greatest concern in the study of behavior. Sixth, "shotgun" mutagenesis is inherently indiscriminate. For every
mutation that is detected and mapped in the offspring of each outlier mouse,
hundreds more exist unknown to the experimenter. While one could eliminate
most extraneous mutations by repeated backcrossing leading to congenic
strains, this is rarely considered in the mouse genome-wide mutagenesis
literature. Seventh, most genes that are monomorphic are so for a reason. For
many if not most such loci, natural selection has eliminated any functional
polymorphisms induced by spontaneous mutations over evolutionary time.
Rendering these polymorphic by chemical mutagenesis often results in a loss
of fitness or even lethality. Among those that are viable, their health may
be compromised. These concerns cause many mutant stocks to be difficult to
maintain and propagate, which increases cost and reduces their utility as
disease models. For the reasons given above, the impressive successes of mutagenesis
screens in dissecting simple traits in simple organisms are likely to be
much more difficult to attain with complex traits (behavior) in genetically
complex organisms (mice). Moreover, the price tag in mice is very high,
especially for complex traits, raising issues of cost-effectiveness compared
to other approaches. Consider also the demands for housing potentially
thousands of new mutant mouse stocks when many animal facilities are already
full to capacity with knockouts and transgenic mice. Finally, natural genetic variation is of interest in its own right in
many biological disciplines, particularly from evolutionary, ecological, and
population genetic perspectives. For example, if we wish to understand the
genetic architecture of a trait in an evolutionary context, naturally
occurring variation is much more likely to provide insightful clues. There
are also immediate biomedical reasons for interest in the large polymorphic
subset of genes for breeding (e.g., selective breeding) of better animal
models of disease states in humans. The demonstrated usefulness of selection
lines for the study of hypertension and alcohol withdrawal severity are
prime examples (Phillips et al., in preparation). Existing variation found
in laboratory stocks of mice and rats are the basis of hundreds of valuable
disease models, and new ones will no doubt continue to be discovered or
developed. More than Just Gene Identification While much of our discussion has focused on gene identification, it is
important to note that this is not the sine qua non of the QTL
approach. Standard practice is to isolate mapped QTLs into congenic strains,
which, when compared to the background strain, allow the study of the
functional effects of the QTL at any desired level of analysis from the
molecular to the organismic. This allows assessment of the effects of a
given QTL on multiple traits (pleiotropy), interactions with other loci (epistasis)
and with environments. This effort to understand QTL effects in a broader
genomic, organismic and environmental context can be quite productive
without knowing the specific gene(s) involved. [Of course, this effort will
be more powerful if the gene has been identified.] Moreover, such studies
will undoubtedly provide important functional clues as to gene identity. A
similar approach could be used for induced mutations as well, but this is
rarely mentioned in the mutagenesis literature. Epistasis is becoming an increasingly important focus of QTL studies
recently, an important new development (e.g., see Hood et al., this volume).
ENU studies do not lend themselves to the study of epistasis as readily
because of secondary effects which compound the difficulty of determining
which interactions are important to the normal organism. The same
situation exists with regard to gene-environment interactions and
correlations, another important consideration for gaining valuable insights
from mouse disease models. Finally, we note that the QTL approach over the past five years has led
to the identification of scores of highly probable candidate genes for many
useful mouse and rat disease models. Most of these would likely not have
been implicated without QTL screens. Thus, QTL studies often provide an
important and powerful hypothesis generating function in contrast to
hypothesis testing. ENU screens also offer this capability. Summary QTL analysis and genome-wide mutagenesis will continue to contribute
greatly to functional genomics in the next decade. Neither approach is an
optimal solution to understanding genetic modulation of complex traits in
mammals. The more we learn about even Mendelian mutations, the more we
appreciate that there are relatively few genuinely simple traits. Epistasis,
genetic background, parental effects, imprinting, and innumerable
gene-environment interactions intrude into originally simple stories. No
matter what the technique, the ride is likely to be "long and bumpy" when
challenges are faced squarely and realistically. The general aim is to
decipher the coordinated actions of many genes and even highly reduced
systems will involve dozens of molecules and dozens of potential exogenous
modulators. In our view, we will need all possible complementary approaches
in functional genomics because the strengths of one will often offset the
weaknesses of another. Given the rapid progress in technology and reagents
that has occurred over the past decade, we are encouraged that the means to
solve, sidestep, or mitigate these problems will be developed. Consider the technology available for QTL work only a decade ago. In
1990, full genome searches were restricted to RFLPs, a cumbersome and
expensive method of genotyping, and software to implement interval mapping
and appropriate Type I error control were not yet available. The first
successful genome-wide search for QTLs in a mouse disease model did not
appear until one year later (Rise et al., 1991). Today, full genome QTL
searches using microsatellite markers are routine, and hundreds of QTLs have
been reported for many valuable phenotypes (Moore and Nagle, 2000; Phillips
et al., in preparation). Readily available software has greatly increased
both the power and accuracy of genome-wide searches (Manly and Olson, 1999).
Higher resolution mapping to 1 cM is now straightforward (Darvasi, 1998),
and sub-cM mapping is beginning to emerge. [These QTL advances have already
greatly enhanced the mapping step of ENU projects.] The availability of full
genome sequence data for four of the most commonly-used inbred strains is
almost at hand. Because of technological advances, both recent and near
future, we are confident that the problems that presently hinder progress
will serve as the instigation for success in the years to come for both the
QTL and mutagenesis approaches. Acknowledgements Work cited as originating in our laboratories was supported by grants
AA10760, AA06243, DA10913, DA05228 and four Department of Veterans Affairs
Merit Review programs. References Belknap, J.K., Metten, P.A., Helms, M.L., O'Toole, L.A., Angeli-Gade, S.,
Crabbe, J.C. and Phillips, T.J. (1993). Quantitative Trait Loci (QTL)
applications to substances of abuse: Physical dependence studies with
nitrous oxide and ethanol. Behav Genet 23:211-220. Belknap, J.K., Dubay, C., Crabbe, J.C. and Buck, K.J. (1997). Mapping
quantitative trait loci for behavioral traits in the mouse. In K. Blum and
E.P. Noble (eds) Handbook of Psychiatric Genetics, CRC Press, Boca Raton,
FL, pp 435-453. Bilbo, S.D. and Nelson, R.J. (2001). Behavioral phenotyping of transgenic
and knockout animals: A cautionary tail. Lab Animal 30:24-29. Brown, SD and Nolan, P. (1998). Mouse mutagenesis--systematic studies of
mammalian gene function. Hum Molec Genet 7:1627-1633. Buck, K.J., Metten, P., Belknap, J.K. and Crabbe, J.C. (1997).
Quantitative trait loci involved in genetic predisposition to acute alcohol
withdrawal in mice., J Neurosci 17:3946-3955. Buck, K.J., Belknap, J.K., Wenger, C., Merrill, C. and Crabbe, J.C.
(1999). Quantitative trait loci involved in genetic predisposition to acute
pentobarbital withdrawal in mice. Mamm Genome 10:431-437. Buck, K.J. and Finn, D. (2001). Genetic factors in addiction: QTL mapping
and candidate gene studies implicate GABAergic genes in alcohol and
barbiturate withdrawal in mice. Addiction 96:139-149. Burmeister, M. (1999). Basic concepts in the study of diseases with
complex genetics. Biol Psychiat 45:522-532. Cook M.N., Williams R.W. and Flaherty L. (2001). Anxiety-related
behaviors in the elevated zero maze are affected by genetic factors and
retinal degeneration. Behav Neurosci 115::468Ð476. Crabbe, J.C., Phillips, T.J., Buck, K., Cunningham, C. and Belknap, J.K.
(1999). Identifying genes for alcohol and drug sensitivity: Recent progress
and future directions. Trends Neurosci 22:173-179. Crawley, J.N. (2000). What’s wrong with my mouse? Behavioral phenotyping
of transgenic and knockout mice. Wiley-Liss, NY, NY. Darvasi, A. (1998) Experimental strategies for the genetic dissection of
complex traits in animal models. Nature Genet 18:19-24. Demarest, C., Koyner, J., McCaughran J., Cipp, L. and Hitzemann R.
(2001). Further characterization and high resolution mapping of
ethanol-induced motor activity. Behav Genet 31:XXX–XXX. Falconer, D.S. and MacKay, T. (1996). Introduction to Quantitative
Genetics, Longman, London. Fehr, C., Belknap, J.K., Crabbe, J.C. and Buck, K.J. (2001). High
resolution mapping of an alcohol withdrawal locus Alcw2 to the
D4Mit80-Mpdz region on mouse chromosome 4. Hamilton B.A., Smith D.J., Mueller K.L., et al. (1997). The vibrator
mutation causes neurodegeneration via reduced expression of PITP alpha:
positional complementation cloning and extragenic suppression. Neuron
18:711-722. Hrabe de Angelis, M. et al. (2000). Genome-wide large-scale production of
mutant mice by ENU mutagenesis. Nature Genet 25:444-447. Lander, E.S. and Schork, N.J. (1994). Genetic dissection of complex
traits. Science 265:2037-2048. Le Roy, I. (2001). Possible causal link between cerebellar patterns of
foliation and hindlimb coordination in laboratory mice: a quantitative trait
loci analysis. Behav Genet 31: XXX-XXX. Lyons, P.A., et al. (2000). Congenic mapping of the Type 1 diabetes
locus, Idd3, to a 780-kb region of mouse chromosome 3: Identification
of a candidate segment of ancestral DNA by haplotype mapping. Genome Res
10:446-453. Manly, K.F. and Olson, J.M. (1999). Overview of QTL mapping software and
introduction to Map Manager QT. Mamm Gen 10:327-334. Malke, H., Steiner, K., Gase, K. and Frank, C. (2000). Expression and
regulation of the streptokinase gene. Methods 21:111-124. Moore, K.J. and Nagle, D.L. (2000). Complex trait analysis in the mouse:
the strengths, the limitations and the promise yet to come. Ann Rev Genet
34:653-686. Mott, R., Talbot, C.J., Turri, M.G., Collins, A.C. and Flint, J. (2000).
A method for fine mapping quantitative trait loci in outbred animal stocks.
Proc Nat’l Acad Sci 97:12649-12654. Nadeau, J.H., Singer, J.B., Matin, A. and Lander, E.S. (2000). Analysing
complex genetic traits with chromosome substitution strains. Nat Gen
24:221-225. Nadeau, J.H. and Frankel, W.N. (2000). The roads from phenotypic
variation to gene discovery: mutagenesis versus QTLs. Nat Gen 25:381-384. Noveroske, J.K., Weber, J.S and Justice, M.J. (2000). The mutagenic
action of N-ethyl-N-nitrosourea in the mouse. Mamm Genome 11:478-483. Nolan, P.M., Kapfhamer, D. and Bucan, M. (1997). Random mutagenesis
screen for dominant behavioral mutations in mice. Methods 13:379-396. Nolan, P.M., et al. (2000). A systematic genome-wide, phenotype-driven
mutagenesis programme for gene function studies in the mouse. Nat Gen
25:440-443. Rise, M. L., Frankel, W. N., Coffin, J. M. and Seyfried, T. N. (1991).
Genes for epilepsy mapped in the mouse. Science 253:669-673. Sandberg, R., et al. (2000). Regional and strain-specific gene expression
mapping in the adult mouse brain. Proc Nat’l Acad Sci 97:11038-11043. Sayah, D.M., Khan, A.H., Gasperoni, T.L. and Smith, D.L. (2000). A
genetic screen for novel behavioral mutations in mice. Mol Psychiat
5:369-377. Schimenti, J. and Bucan, M. (1998). Functional genomics in the mouse:
Phenotype-based mutagenesis screens. Genome Res 8:698-710. Takahashi J.S, Pinto L.H, Vitaterna M.H. (1994). Forward and reverse
genetic approaches to behavior in the mouse. Science 264:1724—1733. Talbot, C.J., Nicod, A., Cherny, S.S., Fulker, D.W. Collins, A.C. and
Flint, J. (1998). High-resolution mapping of a quantitative trait loci in
outbred mice. Nat Gen 21:305-308. Tarantino, L.M., Gould, T.J., Druhan, J.P. and Bucan, M. (2000). Behavior
and mutagenesis screens: the importance of baseline analysis of inbred
strains. Mamm Genome 11:555-564. Tecott, L.H. and Wehner, J.M. (In press). Mouse molecular genetic
technologies: promise for psychiatric research. Arch Gen Psychiat,
58:XXX-XXX. Vitaterna, M.H., King, D.P., Chang, A-M, et al. (1994). Mutagenesis and
mapping of a mouse gene, Clock, essential for circadian behavior.
Science 264:719-725. Wells, C. and Brown, S.D.M. (2000). Genomics meets genetics: towards a
mutant map of the mouse. Mamm Genome 11:472-477. Williams, R.W. (1999). A targeted screen to detect recessive mutations
that have quantitative effects. Mamm Genome 10:734—738. Williams RW (2000)
Mapping genes that modulate brain development: a quantitative genetic
approach. In: Mouse brain development (Goffinet AF, Rakic P, eds).
Springer Verlag, New York, pp 21–49. . Williams, R.W., Airey D.C., Kulkarni A., Zhou G, and Lu, L. (2001).
Genetic dissection of the olfactory bulbs of mice: QTLs on chromosomes 4, 6,
11, and 17 modulate bulb size. Behav Genet 31:61–77. |
Neurogenetics at University of Tennessee Health Science Center
| Print Friendly | Top of Page |
Mouse Brain Library | Related Sites | Complextrait.org
