 |
| |||||
|
| ||||||||
|
| ||||||||
Home  Publications Publications |
|
|
Note to the Reader Text file started Sept 1, 1998. This version updated July 3, 2000. Please refer to: Williams RW (1999) A targeted screen to detect recessive mutations that have quantitative effects. Mammalian Genome 10:734–738.
Print Friendly Abstract. Chromosome substitution strains have been used for
more than four decades to isolate, map, and characterize recessive
mutations in Drosophila. This paper describes an experimental cross
in mice that can be used to define and map induced germ-line mutations
that map to a single chromosome. The cross is a modification and extension
of a conventional three-generation recessive mutagenesis screen. It begins
with an inbred consomic strain that carries the desired target chromosome.
Consomic males are mutated and bred back to the background strain. G1
heterozygous males are also crossed to the background strain. Second
generation male and female progeny that inherited non-recombinant target
chromosomes are identified by genotyping and are bred. The third
generation progeny are genotyped for multiple markers on the target
chromosome, but the aim in this generation is to identify males and
females that are homozygous for the same short interval of the target
chromosome (5 to 25 cM). These near-congenic pairs are bred. Multiple
litters of fourth generation animals that are homozygous for known
segments of the target chromosome are phenotyped. Mutations with
relatively small and somewhat variable effects can be detected and then
mapped to a precision as high as 5–10 cM. The genome is a robust code. Not only do
diploid organisms have two copies of most genes, but many genes also have
closely related variants, or paralogs that serve similar functions (Nadeau
and Sankoff, 1997). Even when both alleles of a given gene are mutated,
the phenotypic repercussions can be subtle. This is what Sewall Wright
(1977) had in mind when he commented that sets of related genes may be
"maintained by selection for quantitative effects, while protected from
drastic effects of inactivation or loss." It follows that an important
problem in a systematic screen for mutations is to devise protocols
capable of detecting more subtle variants. Without sufficient sensitivity,
mutant alleles in a significant fraction of genes may never be detected.
Screens will lack power. In contrast, mutations in genes that lack backup
may be detected many times. To cite an extreme example, a mutagenesis
screen in C. elegans recently produced 63 novel mutations in a single
mechanoreceptor gene (Chalfie and Au, 1989; García-Añoveros and Corey,
1997). In contrast, mutations and knockouts of numerous important genes,
for example, the retinoic acid receptors, often have surprisingly modest
effects that would elude most screens (Luo et al., 1995; Mendelsohn et
al., 1994; Zhou et al., 1998). In this paper I describe a promising
technique designed with this problem in mind. The technique relies on a
novel type of mouse called a consomic mouse and on a four-generation
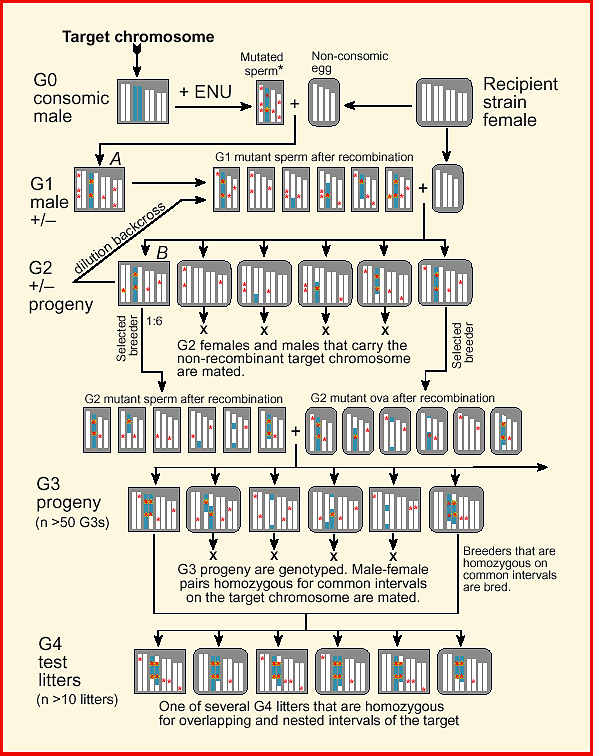
breeding protocol (Fig. 1).
Key features of this method are:
Figure 1.
Download a color high-resolution PDF version of this figure. Methods and MaterialsConsomic StrainThe key starting material for a targeted recessive screen is one or more consomic strains of mice. These strains are produced by introgressing entire chromosomes derived from an inbred donor into an inbred recipient strain (Hudgins et al., 1985). Consomic lines are also referred substitution lines or insertion lines because an exogenous pair of homologous chromosomes is substituted for the usual pair. Several consomic strains have been successfully generated by Jean-Louis Guénet and colleagues (SPRET/Ei chromosomes moved onto a C57BL/6 background; JL Guénet, personal communication). A complete set of consomic strains in which A/J chromosomes have been transferred to a C57BL/6J background are now being generated by Joseph Nadeau, Jonathan Singer, and Eric Lander (1998). Consomic mice in which Chr 7 is derived from A/J have the formal designation C57BL/6J-7A/J (B.A7 for short). I will use the B.A7 strain for illustration in this paper, but any consomic or chromosome substitution strain can be used.
Generating MutationsConsomic males are mutagenized with N-ethyl-N-nitrosourea (ENU) or another mutagen (Fig. 1, top left). Those animals that regain fertility are mated with several females belonging to the recipient strain used to make the consomic line (inbred females, top right). ENU is a potent mutagen of spermatogonia. Each sperm will carry a load of mutations. Tests of specific gene loci suggest that the right dose of ENU can mutagenize up to one gene in a thousand (Russell et al., 1979; 1982; Rinchik et al., 1990). Chr 7 makes up roughly 5% of the mouse genome, and single gametes produced by a B.A7 male will harbor an average of five of the approximately 100 mutations generated randomly across the genome. The great majority of mutations (>90%) carried by the G1 male will be recessives (Kacser and Burns, 1981; Lynch and Walsh, 1998, pp. 63–65).
Generating and Genotyping G2 FemalesFour chromosomes are depicted in Figure 1—the target and three other
chromosomes. The G1 male is a heterozygote at all mutant loci and at all
Chr 7 loci that are polymorphic between A/J and C57BL/6J (left margin).
Like his father, he is mated to the recipient or background strain
(C57BL/6J, in this case) to generate a set of G2 progeny. A majority of
his progeny will inherit recombinant Chr 7s, but a significiant fraction
will inherit a non-recombinant A/J-type chromosome 7 that traces its
descent from a single gamete of the mutated G0 male (e.g., left-most G2
female). For short chromosomes (50 cM) subject to a single obligatory
recombination event during meiosis, two of the four chromatids will
usually be non-recombinant (Lawrie et al., 1995). However, for longer
chromosomes the fraction of non-recombinant chromatids is approximately
0.5L/50, where L is the length of the chromosome in centimorgans. For Chr
7, with a length of approximately 75 cM, the estimated fraction of
non-recombinant chromosome is 0.35, of which half will be derived from the
consomic parental strain. This equation gives estimates that are biased
upward because it fails to account for the non-independence of
recombination events within the tetrad. Interference extends over
approximately 60 Mb (Lawrie et al., 1995) and results in a more uniform
distribution of second crossover events. To sort mice with recombinant and
non-recombinant target chromosomes, all G2 mice are genotyped. The number
of G2 progeny that are genotyped will depend on the length of the target
chromosome, but an average of 30 to 60 G2 animals will typically need to
be tested. To reduce this effort a two step procedure can be employed. All
animals are initially genotyped for markers near proximal and distal ends
of the target chromosome. Mice that are heterozygotes for both markers are
subsequently genotyped at five to ten internal markers spaced at
approximately 10 cM intervals to determine whether or not they inherited
an intact A/J chromosome 7 from their G1 sire. Only those G2 males and
females that are non-recombinant across the entire target chromosome are
retained for breeding.
Genotyping and Breeding G3 progenyNonrecombinant G2 females are crossed to one or more G2 nonrecombinant
males to generate G3 progeny (Fig. 1). All G3s are genotyped at moderate
resolution on the target chromosome (makers spaced at 10 cM). Individuals
homozygous for segments of the target chromosome longer than 5–10 cM are
retained for phenotyping and breeding. An average of one-fourth of G3
progeny will be homozygous for a given part of the target chromosome.
Detecting Lethal MutationsRecessive lethal alleles can make up a large fraction of induced mutations—up to 90% in some screens of Drosophila (Wright, 1977). Unless visible markers are incorporated into a screen these recessive lethals can be difficult to detect. However, in a consomic screen it is easy to recognize and even map recessive lethal mutations. G3 progeny are genotyped at a set of markers that span the target chromosome, and if few or no G3 individuals are homozygous for a particular interval, then this is good evidence that a recessive embryonic lethal mutation has been generated and detected. Similarly, alleles that compromise prenatal and early postnatal viability should lead to segregation distortion of genotypes assayed at weaning. It should subsequently be possible to choose appropriate G3 animals that are heterozygotes across these missing intervals and breed them to confirm, recover, and characterize lethal mutations. Assessing the frequency of missing homozygous intervals among G3 progeny may be an effective way to estimate the efficacy of mutagen dosage.
Phenotyping G4 progenyMatched G3 animals are bred and entire litters of G4 individuals are
phenotyped. All are homozygous across large segments of the target
chromosome. If a segment harbors a recessive mutation, then there should
be a consistent abnormality within a G4 litter. Litters of G4 progeny can
be compared to the original consomic strain and to other litters with
overlapping and non-overlapping regions of homozygosity. How isogenic are G4 test class animals? G4 animals are similar in genetic composition to both the consomic parental strain and to the original inbred recipient strain (Fig. 1). However, even G4 littermates produced from interval-matched G3 parents are not isogenic. First, they carry a variable set of segregating mutations on otherwise inbred non-target chromosomes (Fig. 1). Second, the G4 progeny are genetically heterogeneous in the variable length intervals that flank the homozygous interval of the target chromosome (Fig. 1, bottom). G4 progeny from single litters will therefore have some residual genetic variance. Variance due to off-target mutations can be reduced significantly using an intervening backcross to the wildtype recipient strain (see next section on Collateral Damage). However, the genetic variance within G4 litters should not prevent the detection of mutations. If a mutation maps to the chosen interval of the target chromosome, then other G4 litters that share this interval but that were produced by mating different G3 mice should have the same aberrant phenotype. It is therefore possible to confirm that a mutation maps to the target chromosome and simultaneously refine the position of mutations by comparing several litters from different G3 parents. [Furthermore, by crossing G4 animals to mice carrying small deletions in selected homozygous intervals (Schimenti and Bucan, 1998), it should also be possible to quickly refine the location of a mutation by complementation analysis.] If G4 animals are used to generated additional generations of progeny,
a significant number of these mutations will become fixed. Discussion
Collateral Damage: Are Mutations On-Target?Selective breeding of G2 and G3 progeny enriches for homozygotes on
defined intervals of the target chromosome. However, there will still be
many non-target segments of the genome that harbor mutant alleles (see G3
and G4 progeny in Fig. 1). Mutations that are off-target in G2 progeny are
diluted twofold relative to mutations on the target (compare the mutation
load in the G1 male to that in G2 progeny in figure 1). For this reason,
the probability that a single G3 parent is homozygous for an off-target
mutation originally carried by the G1 male is about 1 in 8. The
probability that two randomly chosen G3 animals will both be homozygous
mutants for off-target intervals is 1 in 64. However, the non-targeted
part of the genome is on average about 20 times larger than the target.
The approximate likelihood that a mutation is off target is therefore
about 1 in 3 (1:64 times 20:1).To compute a more precise estimate would
require information on several factors, including the relative size of the
target chromosome and the homozygous target interval, the relative gene
density of the particular interval, and the relative abundance of lethal
alleles that are off-target. A practical way to assess whether a mutation
is on- or off-target is to examine litters generated from different sets
of G3 parents that share common homozygous intervals. If enough G3 mating
pairs are bred, every part of the target should be represented by two or
more G4 litters. The likelihood that two G4 litters from different parents
will have mutations associated with the same recessive allele on a
non-target chromosome is low—on the order of ~0.005. If three or more
independently derived litters have the same aberration then the mutation
is highly likely to be on-target. Detecting Recessive Mutations in Quantitative Trait LociA key issue in a mutagenesis screen is to have sufficiently sensitive
and economical assays to detect altered traits. Minimizing extraneous
genetic variance is critical. Mackay and colleagues (Anholt et al., 1996)
have used a chromosome substitution protocol and they have been able to
detect and map numerous mutations in Drosophila that affect
behavioral responses to volatile organic compounds. In a consomic screen
the final analysis of deviant phenotypes is based on a comparison between
several litters of G4 progeny and three possible control populations: (1)
the consomic strain, (2) the recipient inbred strain, and (3) non-mutagenized
G4 control lines generated in the same way as the experimental G4 litters
and homozygous for the same interval. Genotyping and Colony Costs of the ScreenGenotyping Costs. A targeted screen using consomic strains
relies on efficient genotyping. Approximately 100 to 200 G2 and G3 animals
will need to be genotyped to characterize on-target mutations carried each
G1 male. If 10 markers were typed per individual, a maximum of 2000
reactions would be required. A more typical estimate is 500 to 1000
reactions. Thanks to the high density of MIT microsatellite markers
(Dietrich et al.,
1992), simple DNA extraction procedures, and streamlined PCR protocols
(Williams and Strom,
<http://www.nervenet.org/papers/PCR.html>), the time, effort, and
expense of genotyping is now comparatively low (~$1/genotype).
Screening for Late-Onset Mutations. It should be practical to retain small numbers of G3 and G4 animals in the colony to study the long-term consequences of novel recessive alleles. The original G3 mating pairs will often be 100-days-old before the analysis of their first litter of G4 progeny is complete. If no interesting phenotypes are initially detected in the G4 animals, then it may nonetheless be useful to hold several sets of G3 or G4 breeders in anticipation of possible late-onset traits. It may often be possible to test an entire chromosome by retaining only animals with long congenic intervals. In the absence of recessive lethal alleles, as few as one to five pairs of mice could cover an entire chromosome. Periodic test matings may be useful to ensure that mutations with late lethal effects are not lost.
Targeted Screens—Disadvantages and AdvantagesA targeted screen ignores mutations generated across most of the
genome—19 out of 20 mutations will be rejected in a typical consomic
screen. For this reason, a targeted approach may not be appropriate (1) if
the screening procedure is tightly focused, reliable, rapid, and
inexpensive; (2) if there is good evidence that traits of interest are
known to be controlled by small numbers of loci; and (3) if mapping
mutations is a low priority and one that can be more conveniently done
after mutations have been identified. One particular potential problem
with a targeted approach is the possibility that a narrow chromosomal
focus will be coupled with a low yield of mutations (Schimenti and Bucan,
1998). The consomic approach enriches for homozygotes and this does
increase the efficiency and sensitivity of the screen. However, even after
selectively breeding G2 and G3 individuals, some mutations that do not map
on the target chromosome will be detected among G4 progeny. In order to
retain focus on mutations that can be rapidly mapped and for which a list
of candidate genes can be identified, these off-target mutations will
generally need to be ignored. Whether this focused approach yields a
sufficiently high output of novel recessive alleles will depend in part on
the ingenuity, sensitivity, and number of tests that can be performed on
G4 litters. Using Congenic Strains?A screen could in principle be adapted to exploit smaller target intervals that have been isolated in congenic, rather than consomic, strains of mice. In congenic strains the length of the donor interval is a function of the number of backcrosses, but even after ten backcross generations, the donor interval is typically 20 cM in length (~200/n cM, where n is the number of generations of successive backcrosses). One disadvantage of using congenic strains is that a significant fraction of mutations will occur in linked intervals that flank the congenic segment. These adjoining intervals will be larger than the congenic interval itself. This will make mapping mutations generated in congenic strains somewhat more unpredicable than those generated using consomic strains.
Precedents: Other Targeted MethodsTargeted screens for recessive mutations have been carried out for many
years and it is important to compare the consomic design with well known
alternatives. One of the first and best protocols to screen for recessive
mutations takes advantage of hemizygous lines that are missing all or part
of particular chromosomes (Muller, 1927; Justice et al., 1997, Schimenti
and Bucan, 1998). Muller's classic mutagenesis experiments exploited the
hemizygosity of the X chromosome of male fruit flies. But artificial
hemizygous intervals can now be generated in mice of both sexes by
deleting a short segments from one autosome. Mice from which 5 to 15 cM
has been deleted are usually viable (Justice et al., 1997; Schimenti and
Bucan, 1998). Recessive alleles that are in trans to the deletions are
unmasked and mice have phenotypes similar, if not identical, to those of
homozygote mutants. To expose a recessive mutation involves breeding G1
heterozygote carriers like those in Figure 1 to deletion stock. An average
of one in four progeny of such a cross will be a hemizygous mutant.
Rinchik and colleagues used mice from which the interval on chromosome 7
extending from tyrosinase to shaker 1 had been deleted (Rinchik et al.
1990; Rinchik, 1991). G1 females carrying mutations inherited from
ENU-mutagenized sires were crossed to males hemizygous for the deletion.
By a clever choice of coat-color mutations, they were able to restrict
their main analysis to the hemizygous G2 test class animals. Schimenti and
Bucan (1998) review a G2 screen in which recombination in a deletion
interval is suppressed by using a stock that carries a large inversion and
a coat-color mutation. This refinement simplifies the analysis by forcing
a more nearly perfect concordance between visible markers and genotype
(see their figure 3).
Drosophila Precedents. The consomic procedure outline here also
has precedents that trace back to a series of experiments by Dobzhansky
and colleagues (e.g., Dobzhansky et al., 1959) in which single chromosomes
of wild populations of fruit flies were introgressed into special
laboratory strains to examine the load of recessive lethal alleles carried
by natural populations. Recombination in fruit flies can be suppressed,
and given a laboratory strain with the right visible markers it is
relatively simple to follow the assortment of chromosomes in a three
generation cross. In several impressive studies, Mackay and colleagues
(Mackay et al., 1992; Lyman et al. 1996; Anholt et al., 1996) have
exploited chromosome substitution techniques to study effects of induced
mutations. Mutated chromosomes were introgressed into isogenic backgrounds
and the effects on fecundity, sensory bristle number, and olfactory
behavior were assayed. In these studies a complex breeding scheme
exploiting visible markers and tagged P-elements (Fig. 1 of Lyman et al.,
1996) was used to filter progeny and generate test class animals (G4
through G6). They then examined large numbers of nearly isogenic mutants
to detect subtle QTL-like effects. P-elements were subsequently mapped at
high-resolution by in situ hybridization. In a consomic screen using mice,
visible markers are replaced by molecular markers made visible using
simple PCR protocols. There are many specific differences between the
Drosophila substitution mutagenesis screen (see Fig. 1 of Lyman et
al., 1996) and the consomic procedure described here, but the key goals
are sharedÑminimize extraneous genetic variance while maximizing the
efficiency with which induced mutations can be mapped. As in
Drosophila, there is still extraneous mutational variance in the mouse
consomic screen. The source of this variance in flies is not yet clearly
understood (Lyman et al., 1996), but in consomic mice most of this
variance will be associated with the potentially large load of off-target
mutations. These mutations can only be removed by repeatedly backcrossing
G2 non-recombinants to the inbred recipient strain. AcknowledgementsThis research was supported by grant RO1 EY06627 from the National Eye
Institute and RO1 NS35485 from the National Institute of Neurological
Disorders and Stroke. I thank Kathryn Graehl for helpful discussion and
for editing the text and Figure 1. My thank to Drs. Rosemary Elliott,
Lorraine A. Flaherty, Jean-Louis Guénet, Eugene Rinchik, John Schimenti,
Benjamin Taylor, and David W. Threadgill for comments and corrections. References
Anholt RRH, Lyman RF, Mackay TFC (1996) Effects of single P-element insertions on olfactory behavior in Drosophila melanogaster. Genetics 143:293–301. Brown SDM, Peters J (1996) Combining mutagenesis and genomics in the mouse—closing the phenotype gap. Trends Genet 12:433–435. Brown SDM, Nolan PM (1998) Mouse mutagenesis-systematic studies of mammalian gene function. Hum Mol Genet 7:1627–1633. Chalfie M, Au M (1989) Genetic control of differentiation of the Caenorhabditis elegans touch receptor neurons. Science 243:10273–1033. Dietrich W, Katz H, Lincoln SE, Shin HS, Friedman J, Dracopoli NC, Lander ES (1992) A genetic map of the mouse suitable for typing intraspecific crosses. Genetics 131:423–447. <http://carbon.wi.mit.edu:8000/cgi-bin/mouse/index> Dohzhansky T, Spassky B (1954) Genetics of natural populations. XXII. A comparison of concealed variability in Drosophila prosaltans with that in other species. Genetics 39:472–487. Dohzhansky T, Levene H, Spassky G, Spassky N (1959) Release of genetic variability through recombination. III. Drosophila prosaltans. Genetics 44:75–92. García-Añoveros J, Corey DP (1997) The molecules of mechanosensation. Annu Rev Neurosci 20:567Ð594. Hudgins CC et al., (1985) Studies of consomic mice bearing the Y chromosome of the BXSB mouse. J Immunol 134:3849–3854. Hrabé de Angelis M, Balling R (1998) Large scale ENU screens in the mouse. Genetics meets genomics. Mutation Research 400: 25–32. Justice M (1998) Mouse germline mutagenesis. In: Mouse genetics and transgenics: a practical approach (Jackson I, Abbott C, eds). pp XXX-XXX. New York: Oxford UP. Justice MJ, Zheng B, Woychik RP, Bradley A (1997) Using targeted large deletions and high-efficiency N-ethyl-N-nitrosourea mutagenesis for functional analyses of the mammalian genome. Methods 13:423–436. Kacser H, Burns JA (1981) The molecular basis of dominance. Genetics 97:639–666. Kasarskis A, Manova K, Anderson KV (1998) A phenotype-based screen for embryonic lethal mutations in the mouse. Proc Natl Acad Sci 95:7485–7490. Lawrence PA (1992) The making of a fly. London: Blackwell. Lawrie NM, Tease C, Hulten MA (1995) Chiasma frequency, distribution and interference maps of mouse autosomes. Chromosoma 104:308–314. Lufkin T, Lohnes D, Mark M, Dierich A, Forry P, Gaub MP, LeMeur M, Chambon P (1993) High postnatal lethality and testis degeneration in retinoic acid receptor alpha mutant mice. Proc Natl Acad Sci USA 90:7225–7229. Luo J, Pasceri P, Conlon RA, Rossant J, Giguere V (1995) Mice lacking all isoforms of retinoic acid receptor beta develop normally and are susceptible to the teratogenic effects of retinoic acid. Mech Dev 53:61–71. Lyman RF, Lawrence F, Nuzhdin SV, Mackay TFC (1996) Effects of single P element insertions on bristle number and viability in Drosophila melanogaster. Genetics 143:277–292. Lynch M, Walsh B (1998) Genetics and analysis of quantitative traits. Sinauer, MA. Marker PC, Seung K, Blande AE, Russell LP, Kingsley DM (1997) Spectrum of Bmp5 mutations from germline mutagenesis experiments in mice. Genetics 145:435-443. Mendelsohn C, Lohnes D, Decimo D, Lufkin T, LeMeur M, Chambon P, Mark M (1994) Function of the retinoic acid receptors (RARs) during development (II). Multiple abnormalities at various stages of organogenesis in RAR double mutants. Development 120:2749–2771. Morel L, Mohan C, Yu Y, Croker BP, Tian N, Deng A, Wakeland EK (1997) Functional dissection of systemic lupus erythematosus using congenic mouse strains. J Immuno 158:6019–6028. Muller HJ (1927) Artificial transmutation of the gene. Science 46:84–87. Nadeau JH, Sankoff D (1997) Comparable rates of gene loss and functional divergence after genome duplications early in vertebrate evolution. Genetics 147:1259–1266. Nolan PM, Kapfhamer D, Bucan M (1997) Procedures for the identification of dominant behavioral mutations in mice using ENU mutagenesis. I: Methods: A companion to methods in enzymology 13:379–395. Pickard GE, Sollars PJ, Rinchik EM, Nolan PM, Bucan M (1995) Mutagenesis and behavioral screening for altered circadian activity identifies the mouse mutant, Wheels. Brain Res 705:255–266. Rinchik EM, Carpenter DA, Selby PB (1990) A strategy for fine-structure functional analysis of a 6– to 11–centimorgan region of mouse chromosome 7 by high-efficiency mutagenesis. Proc Nat Acad Sci USA 87:896–900. Rinchik EM (1991) Chemical mutagenesis and fine-structure functional analysis of the mouse genome. Trends Genet 7:15–21. Russell WL, Kelly PR, Hunsicker PR, Bangham JW, Maddux SC, Phipps EL (1979) Specific-locus test shows ethylnitrosourea to be the most potent mutagen in the mouse. Proc. Nat. Acad. Sci. USA 76: 5918–5922. Russell WL, Hunsicker PR, Raymer GD, Steele MH, Stelzner KF, Thompson HM (1982) Dose response curve for ethylnitrosurea specific-locus mutations in mouse spermatogonia. Proc Natl Acad Sci USA 79: 3589–3591. Schimenti J, Bucan M (1998) Functional genomics in the mouse: phenotype-based mutagenesis screens. Genome Res 8:698Ð710. Shedlovsky A, King T, Dove W (1988) Saturation germ line mutagenesis of the murine t-region including a lethal allele at the quaking locus. Proc Natl Acad Scie USA 85:180-184. Suda Y, Matsuo I, Aizawa S (1997) Cooperation between Otx1 and Otx2 genes in developmental patterning of rostral brain. Mech Dev 69:125–141. Vogel E, Natarajan AT (1979) The relatiion between reaction kinetics and mutagenic action of mono-functional alkylating agents in higher eukaryotic systems. I. Recessive lethal mutations and translocations in Drosophila. Mutation Res 62:51Ð100. Williams RW (1998) Neuroscience meets quantitative genetics: using morphometric data to map genes that modulate CNS architecture. The 1998 Short Course in Quantitative Neuroanatomy (Morisson J, Hof P, eds) pp. 66-78. Washington: Society for Neuroscience. (http://www.nervenet.org/papers/ShortCourse98.html). Wright S (1977) Evolution and genetics of populations, Vol 3. Chicago: Univ Chicago Press. You Y, Bergstrom R, Klemm M, Lederman B, Nelson H, Ticknor C, Jaenish R, Schimenti JC (1997a) Chromosomal deletion complexes in mice by radiation of embryonic stem cells. Nat Genet 15:285–288. You Y, Browning VL, Schimenti, JC (1997b) Generation of radiation-induced deletion complexes in the mouse genome using embryonic stem cells. Methods 13:409–421. Zhou G, Strom RC, Giguere V, Williams RW (1998) Modulation of retinal
cell populations and eye size in retinoic acid receptor knockout mice. Soc
Neurosci Abst 24:1033. Related Links
Since 4 Sept 98
|
Neurogenetics at University of Tennessee Health Science Center
| Print Friendly | Top of Page |
Mouse Brain Library | Related Sites | Complextrait.org
